
上海自贸区进口医疗器械贴标展开深入讨论 ▎上海生物医药领域地方立法研讨会议召开

2024年5月17日,“中国(上海)自贸试验区进口医疗器械加贴中文标签暨医疗器械地方立法讨论会”在上海浦东软件园【SITSW / 中国•上海国际贸易单一窗口】/亿通国际报告厅举行。
进一步加速上海生物医药先导性产业发展,贯彻国务院关于《全面对接国际高标准经贸规则推进中国(上海)自由贸易试验区高水平制度型开放总体方案》,深化改革开放,推动具体政策措施落地实施是本次会议的主题。
会议围绕起草本市生物地方性法规的立法任务,针对促进进口医疗器械本地化生产;解决上海研发型企业高效获得国际相关研发物品资源支持;促进自贸区在医疗器械领域采用国际通行规则实现标签文种转化等三个问题开展了交流。
上海市药品监督管理局领导带领政策法规部门立法相关同志出席了会议。上海自贸区内五十多家著名医疗器械跨国公司的供应链管理部门领导近100多人出席会议并做了大会发言和交流。

一、扩大上海自贸区进口医疗器械保税区贴标讨论
去年底国务院下达《全面对接国际高标准经贸规则推进中国(上海)自由贸易试验区高水平制度型开放总体方案》,方案第11条提出达到条件的进口医疗器械其主体可以在上海自贸区内进一步扩大试点对进口医疗器械贴标。
随着改革开放力度加强,各地开展国际贸易都有同样需求。如何做到进一步与国际接轨,在高标准制度化条件下,逐步与全球自贸区形成等同的国际贸易环境,促进RCEP对接和实施,促进“一带一路”国家经济策略发展,本身就是深化国际开放创新监管的问题。
会议发言指出,尽管上海外高桥在2013年中国建立第一个自贸区过程中,实施进口医疗器械标签国别/文种转换的成果已经具备十年成熟经验,当前在2021年【739条例】上位法顶层约束下,讨论这个问题仍然是制度创新的挑战。简单说【739条例】没有解释国家特殊经济监管区内如何监管医疗器械,境内关外特殊区域也没现成文字解释【739条例】应该如何执法。

【对标国际自贸区如何监管医疗器械才能实现安全有效】
本次会议前,上海市药监局提出了进口医疗器械贴标规定的文件(征求意见稿),并在市府一网通办征求各方意见。方案形成前,召开过多次企业讨论会议,与海关监管部门多次沟通。出席会议的几十家企业,带到会上81条具体意见建议,大部分是关于操作质控的意见,其中一些涉及保税区监管的核心问题,需要引起注意。汇总归纳如下:
- 这次改革探索的前提是进一步高标准对标国际贸易环境,自贸区/保税区是境内关外特殊监管区,各国可以在区内设立海外加工地,承接海外标签加工是否受到条例许可证制约,区内进口保税货物仓库转移是正常业务状态;上海保税区发展国际港口货物分拨中心,转口、进口货物标签文种转换是基本业务活动,在保税状态产品英文标签转换中文,或者是其他国别文种都是常态业务。
- 医疗器械产品的标签加工是什么行为,是生产行为还是经营行为?标签转换加工业务活动的性质和质量管控的途径不能脱离全球法规和国内条例的定义,存在模棱两可和各自理解空间,最终会导致现场实际操作和监督困难;
- 在中国境内实施标签加工的主体责任人是谁?进口注册证的持证主体在境外没有完成最终标签生产加工,现在继续到其他语言国家(例如新加坡)的境内关外地区进行生产加工,原来注册证的第一责任人(Manufacturer )的主体需要更换为总代理 ?与【条例】已经规定的顶层结构是否有冲突;进口医疗器械在国内的责任主体现在需要变成二个主体承担?如果总代理负责建立质量体系实施标签加工转换,那么进口产品释放主体是谁?需要形成由第二责任主体/总代理负责进口产品释放吗?这样对【条例】顶层监督结构产生修正。
【条例向下形成进口产品二个责任主体的决策需要十分慎重】
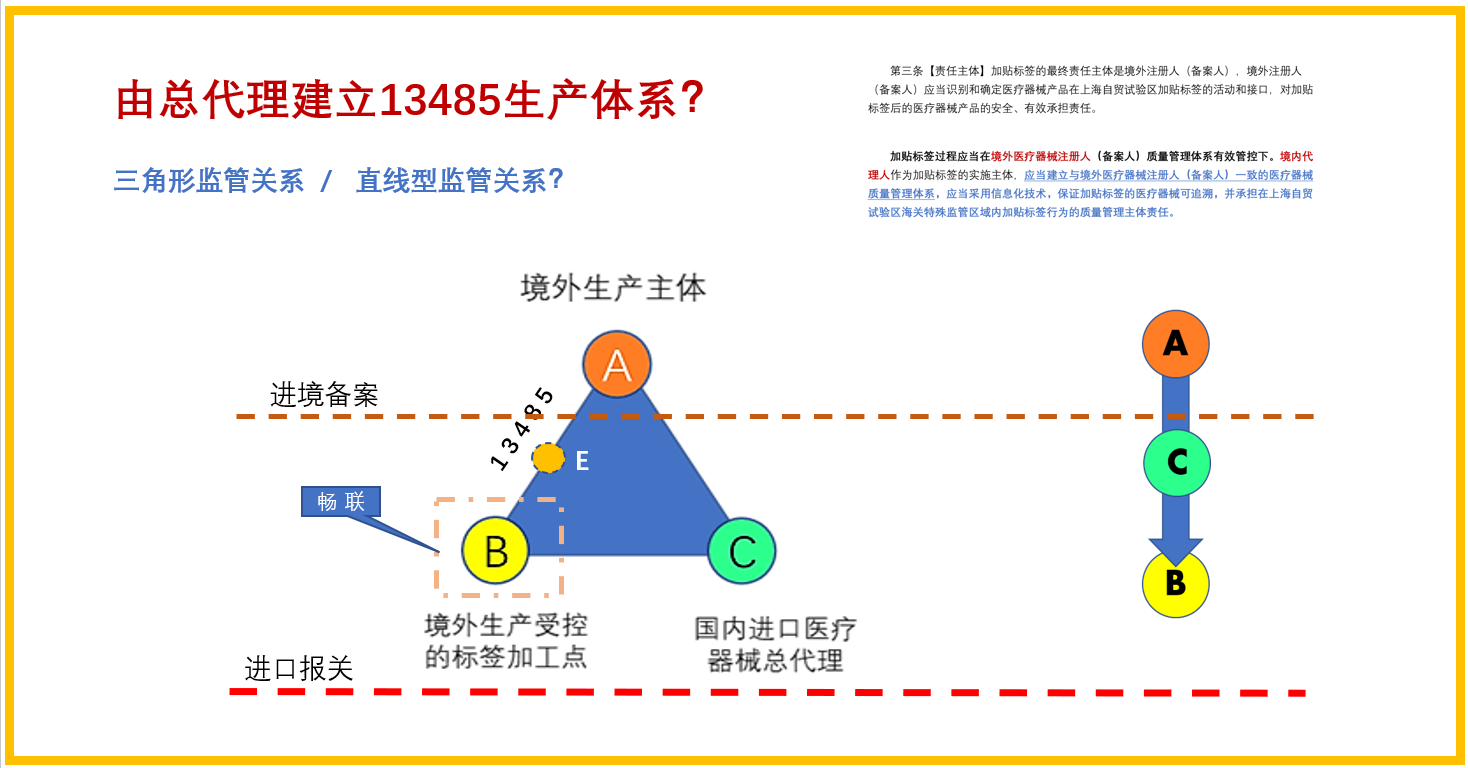
境外的注册人/备案人在中国的总代理的责任地位在条例是清晰的。简单概括为:【总代理是医疗器械境外注册人/备案人承诺履行中国《医疗器械监督管理条例》规定的一切法定责任,并在中国境内指定的具有法人资格的全责受托人】。如果延申讨论一下,【条例】向下,需要对总代理另外建立法规吗,需要把责任一条一条写清楚,能写得清楚 ?
- ISO-13485 认证检查,全球实施专业人员检查,医疗器械产品复杂,如果每条检查要求写出来,估计倒是适得其反,检查漏洞产生了。那么标签加工如果采用生产质量体系管理,需要在实施规定中具体写出体系检查的每一条具体要求吗?罗列出来的操作要求比13485标准的条款更有效吗?(征求意见几乎有十多条具体要求/GSP 写法);境外产品质量体系检查/认证归口国家层面统一监管,如果其中有一段生产体系发生在其他国家,例如标签体系发生在境内关外实施,境内的属地监管人员可以主动发起检查/认证吗?
- 从监管角度,进口医疗器械有证产品在自贸区的境内关外地区建立标签海外加工点,是采用备案管理还是许可管理,如果是备案管理,应该如何核对资料准确性?如果生产过程是采用国际体系认证,境内外分段体系衔接,为了事后监管延续,这一个过程监管人员需要参加吗,有机会参加吗,需要用什么身份参加,备案资料向谁递交?时限、程序;与属地海关平行监管从备案提交就开始,资料分享信息节点在哪里等等,二个监管部门协调监管原则应该在规定中明确。
本次大会议程有五位跨国公司总代理代表对自贸区进口的医疗器械标签加工转换问题做了发言。’提出讨论的建议和意见包括:国际贸易环境高标准对接的标准、定义;标签加工实施主体;标签加工体系和境内外对接;监管等四大类。十年来原外高桥进口标签加工转换,在第一个自贸区的“先行先试”,形成了完整的监管法理依据和实际管控经验,会议认为不应该推倒重来。
提交的意见最集中的是签加工需要报告的条款。会议交流介绍,有的企业每月十几个集装箱,上百万标签实施,生产过程经常有内部合规管控的变更情况,需要企业报告什么、如何报告,报告的数据监管部门如何利用等具体问题,如果采用13485国际认证管理,这些条款是否需要编写进入规定(甚至删除)应该认真研究,标签加工规定全文应该着重解决上述管理原则。
二、国家药监104公告境外医疗器械产品转国内生产
会议过程还交流了2020年国家局104号公告,关于进口医疗器械产品转国内生产的立法问题。会议发言代表提出,按照条例框架,一个与已经上市产品相类似的产品批准释放,全球FDA不会采纳非技术性标准进行评估。被转让企业“血统纯净度“问题,转移境外产品核心技术的占比等等,是国家经济工作会议总方针贯彻问题,不应该在本次立法中在属地范围内争论。相似产品上市释放评价问题,应该采用全球医疗器械风险评价和标准体系应用的原则来考虑。会上德国蔡司提供的欧盟“实质性等同 /Substantial Equivalence /SE ”原则处理相似产品释放值得进一步学习和研究。
会议同时对第三个专题《科创物资进口“白名单制度”高效实施》展开进行讨论。已经发布了会议情况,不在此累述。具体见:https://mp.weixin.qq.com/s/m08_SLelYQ8bvGnQACGBNA
本次会议是一次有效率的会议。各位代表提出的建议和意见在下一步形成立地方立法的起草中有重要参考价值。当前医疗器械地方立法行业关注:自贸区扩大进口医疗器械标签加工;104号公告的实施;科创物资快速进口等监管原则等三个方面是行业的迫切,会议希望继续深入开展探讨,MDTA 将继续搭建好企业和政府监管的高效交流平台。
【会场图片花絮】
.png)
.png)
.png)
.png)

.png)






【以上图片来源于泓明媒体组协助】
联系方式
地址:中国(上海)自由贸易试验区 外高桥美约路222号五楼501室
邮 编:200131
传 真:58661522
电 话:58661516
邮 箱:mdta@mdta.org.cn
下载专区
相关链接


©2018 上海浦东医疗器械贸易行业协会
沪ICP备19039930号-1 沪公网安备31019002000210号 互联网药品信息服务资格证书编号:(沪)-非经营性-2022-0001
技术支持:维程教育
 loading......
loading......